Feature finding with MZmine2¶
Please follow this (link) to install the software and dependencies.
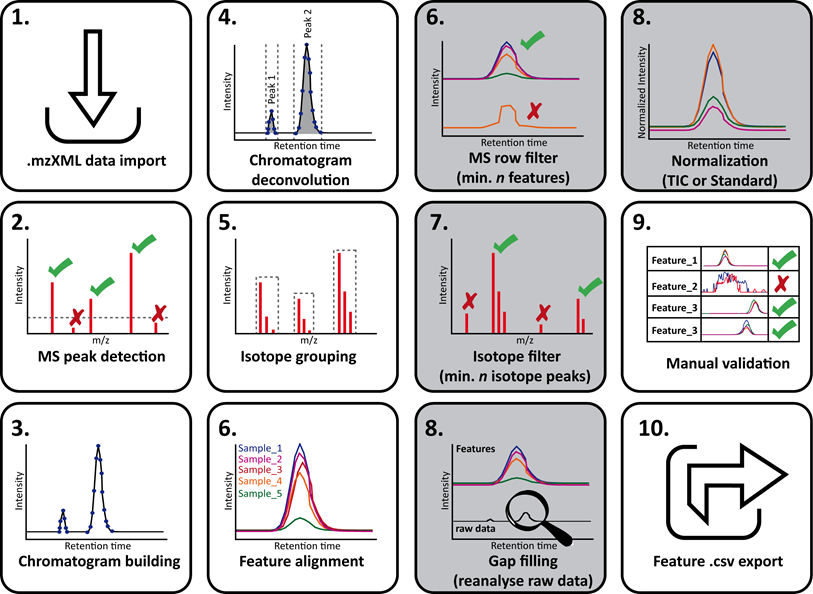
Complete workflow view¶
1. Start mzMine2¶
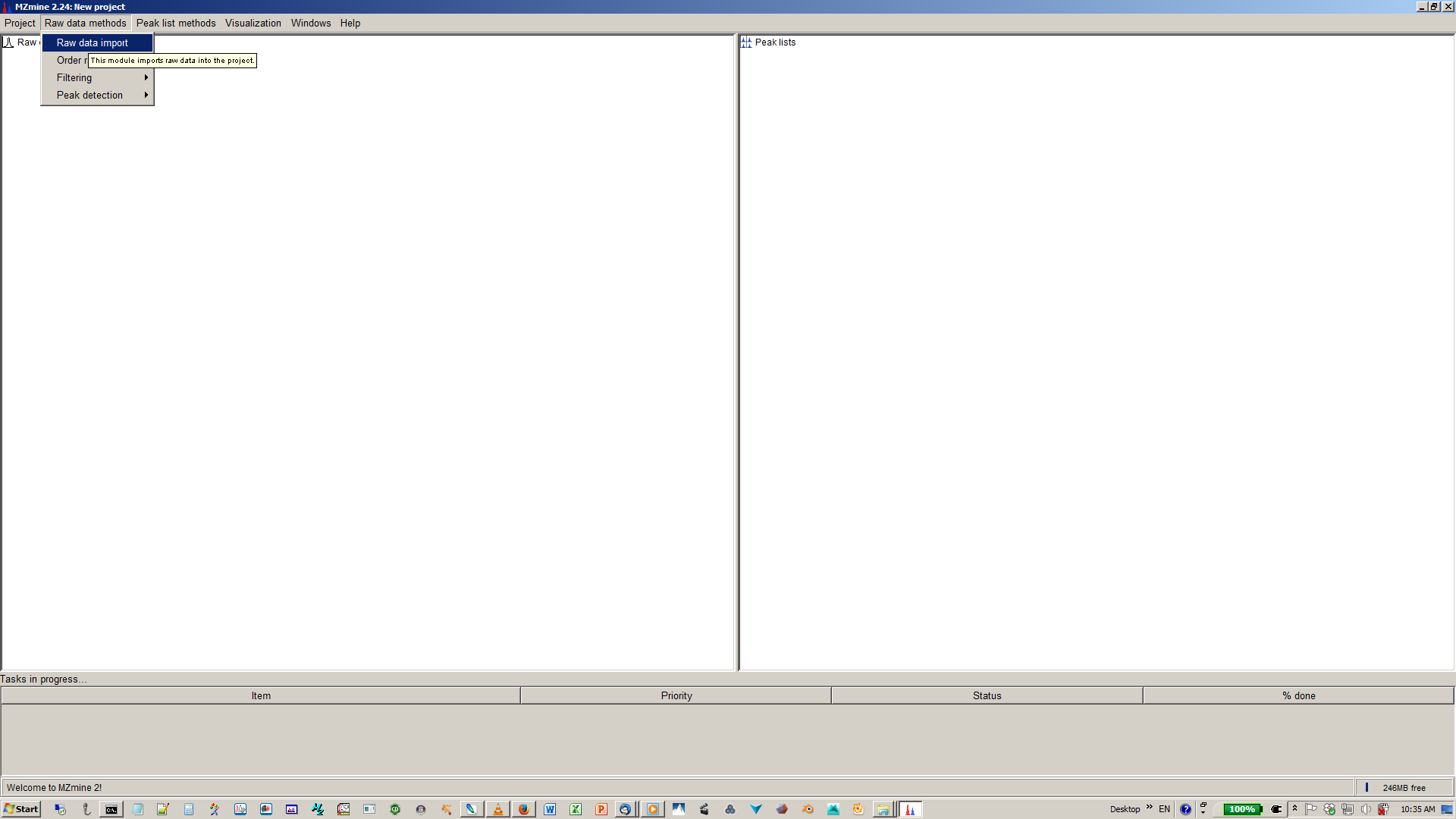
2. Click on raw data import in drop down menu and select .mzxml files¶
3. Click on mass detection in drop down menu¶
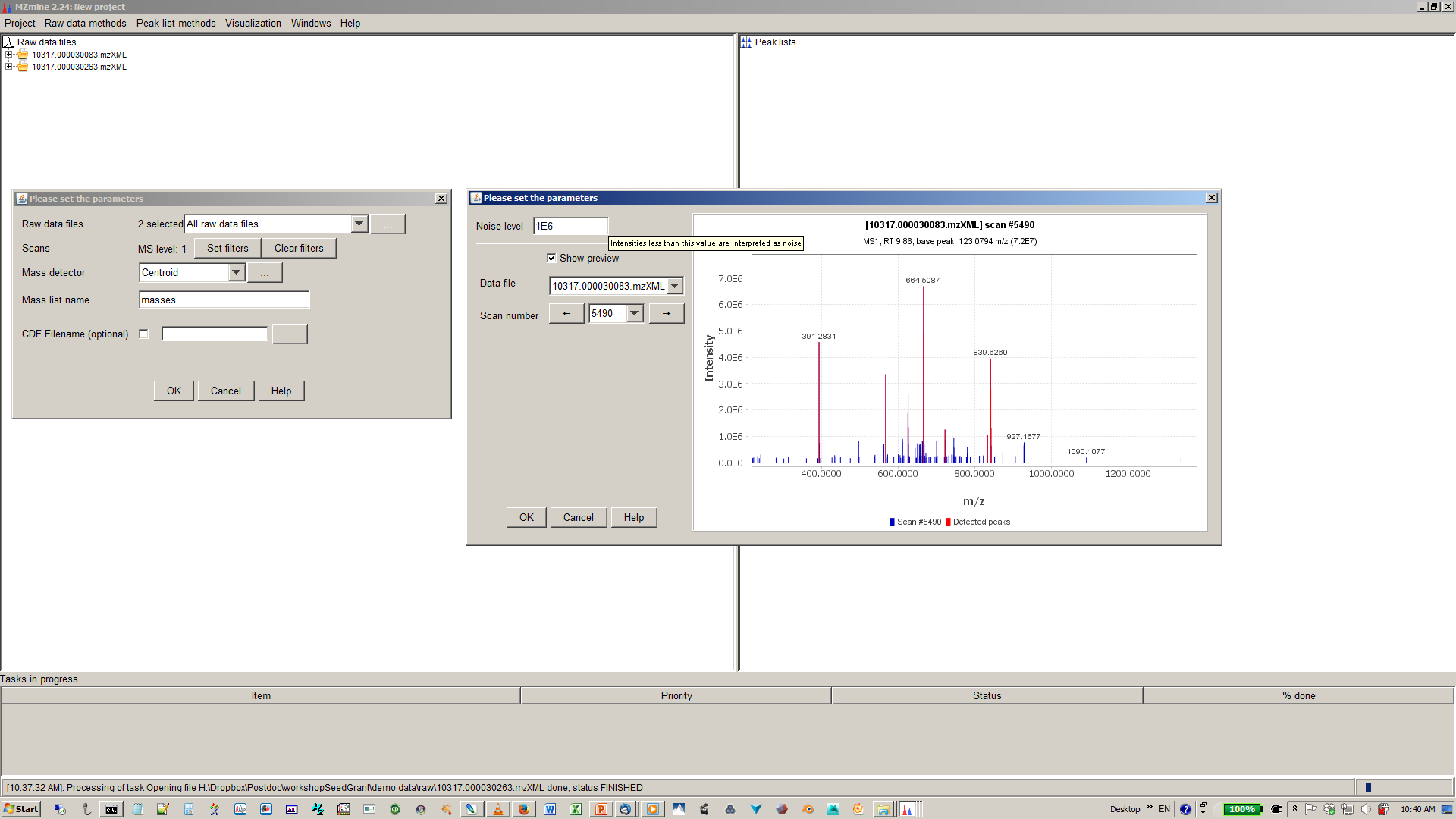
4. Specify intensity cut-off and mass list¶
5. Build XICs with chromatogram builder¶
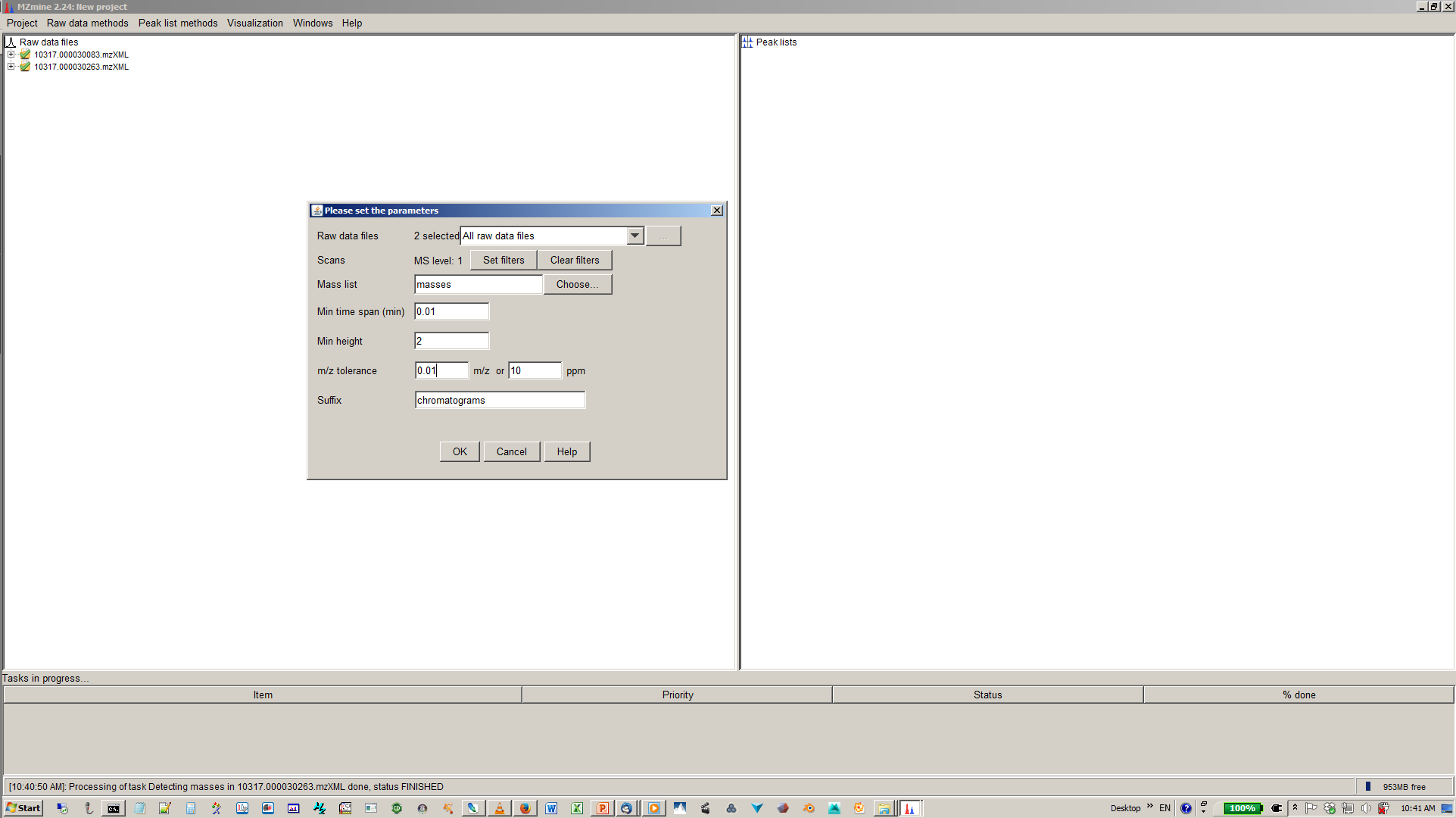
6. Specify mass list, mass tolerance min. time span and min. hight¶
7. Deconvolute isobaric peaks with chromatogram deconvolution¶
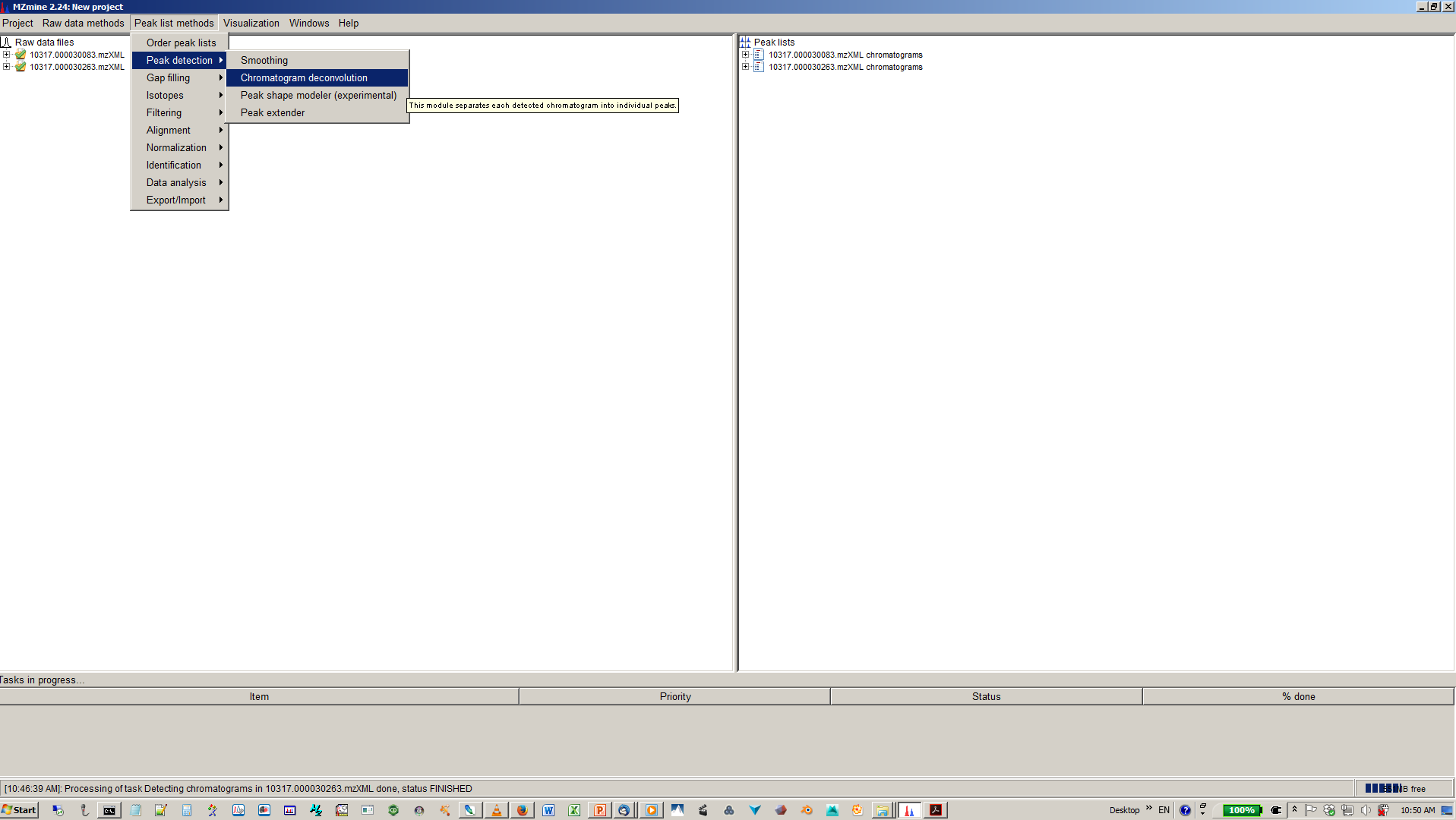
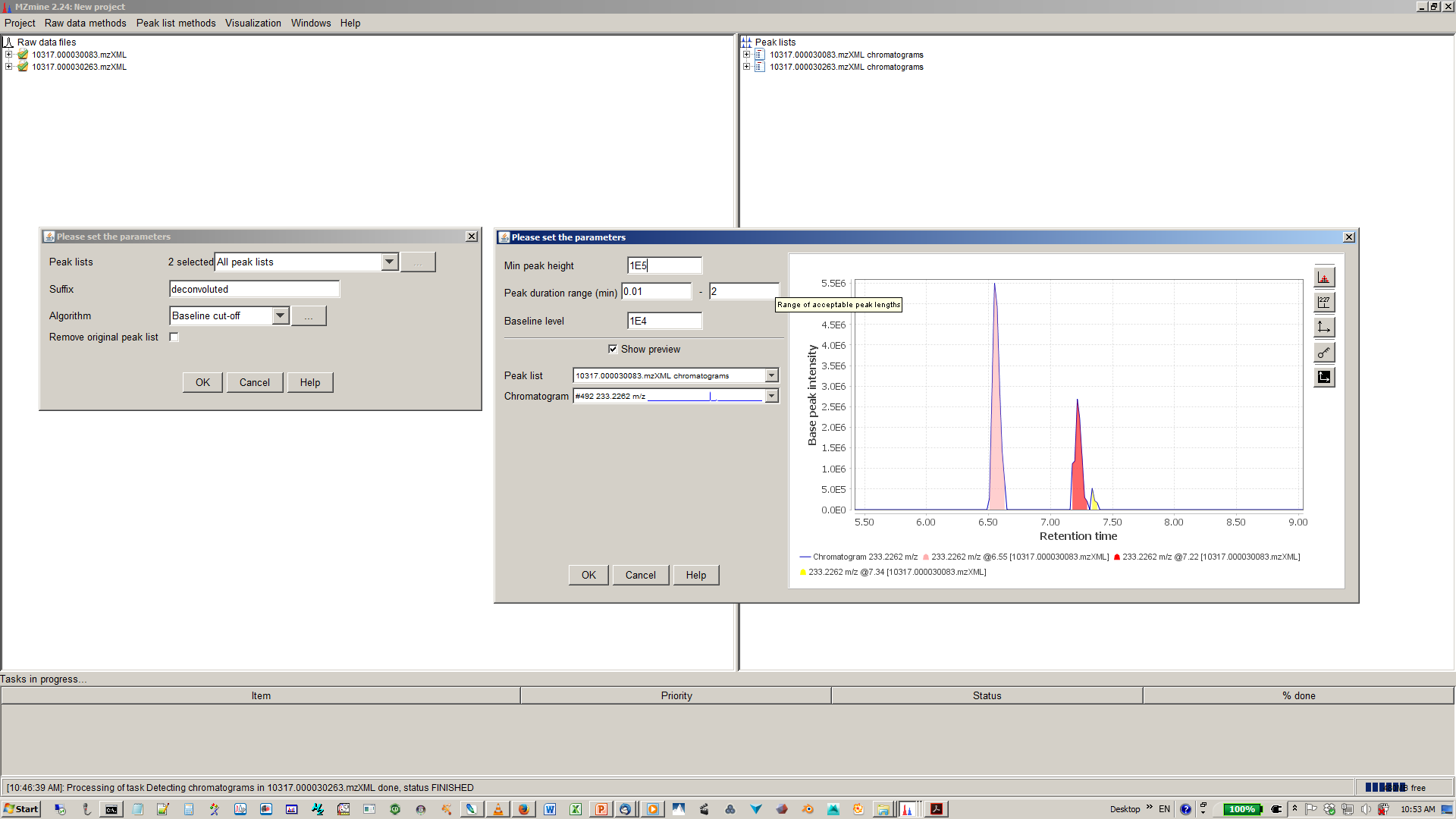
8. Specify algorithm (base line cut-off or local minimum search and parmaters¶
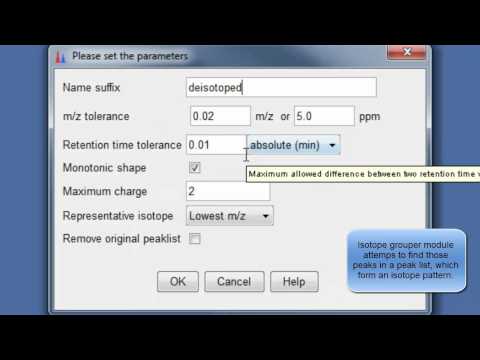
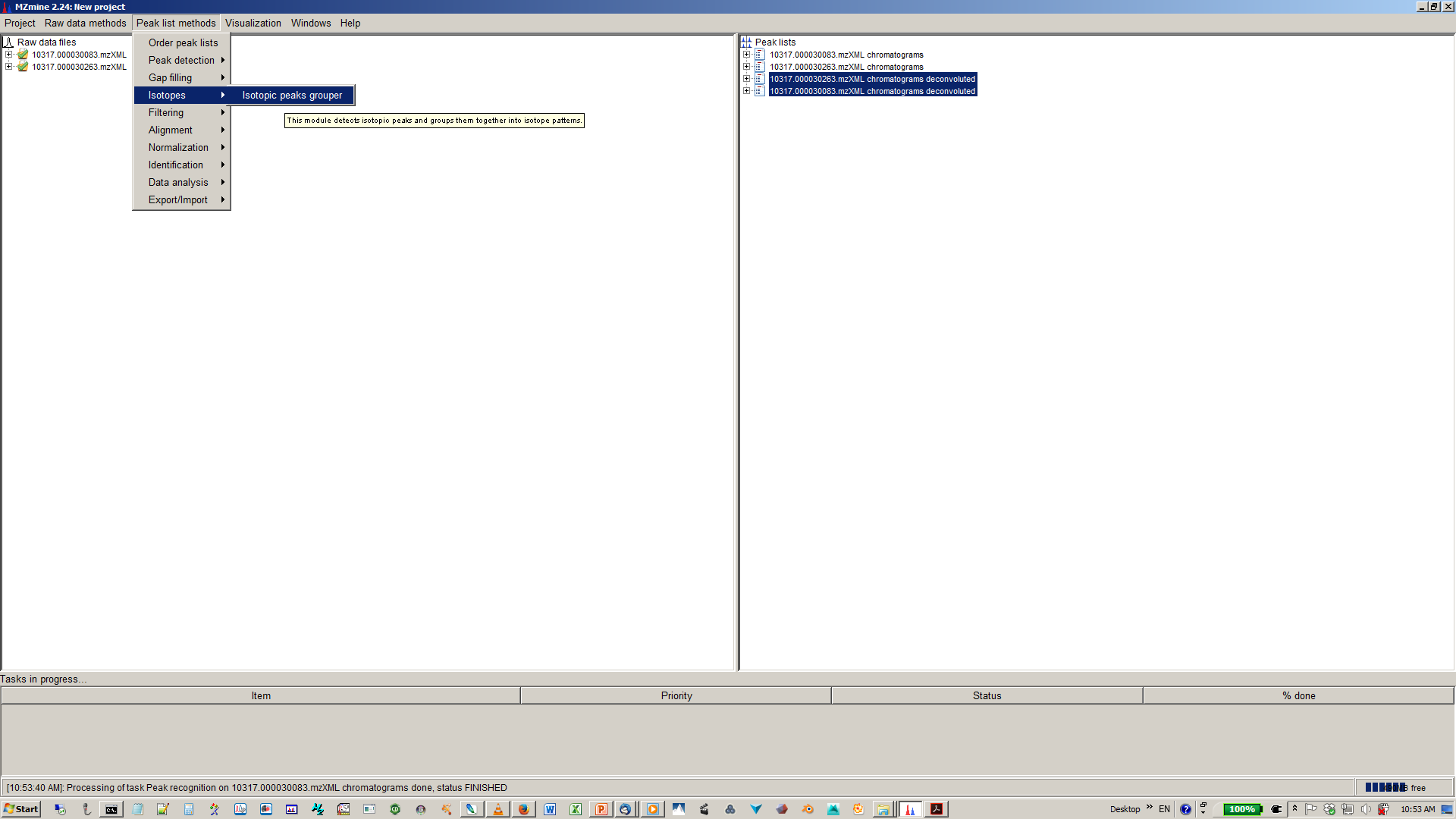
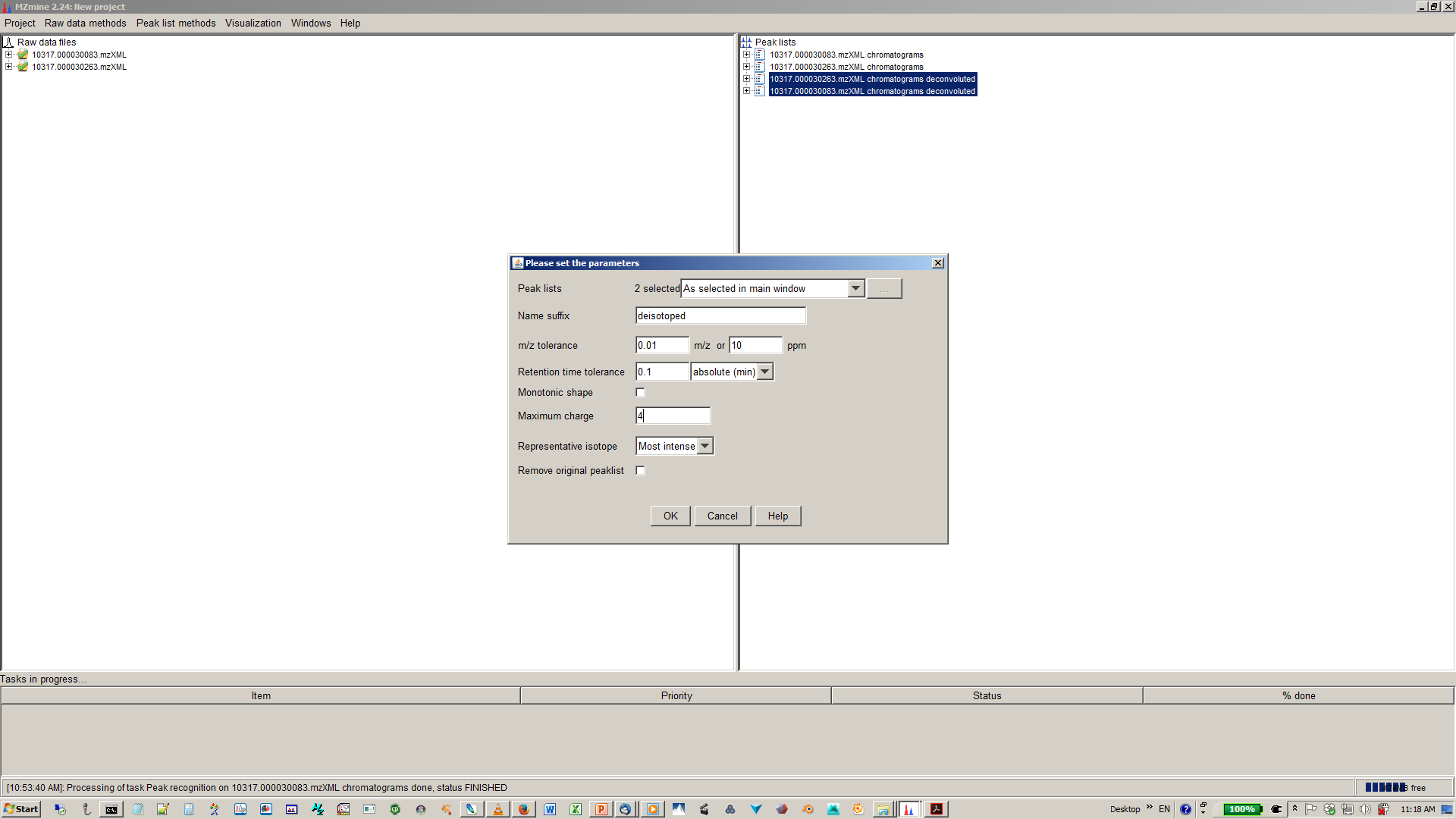
9. Perform deisotopization through isotope peak grouper¶
10. Specify parameters for isotope peak grouping¶
11. Align XICs from different sample to one matrix¶
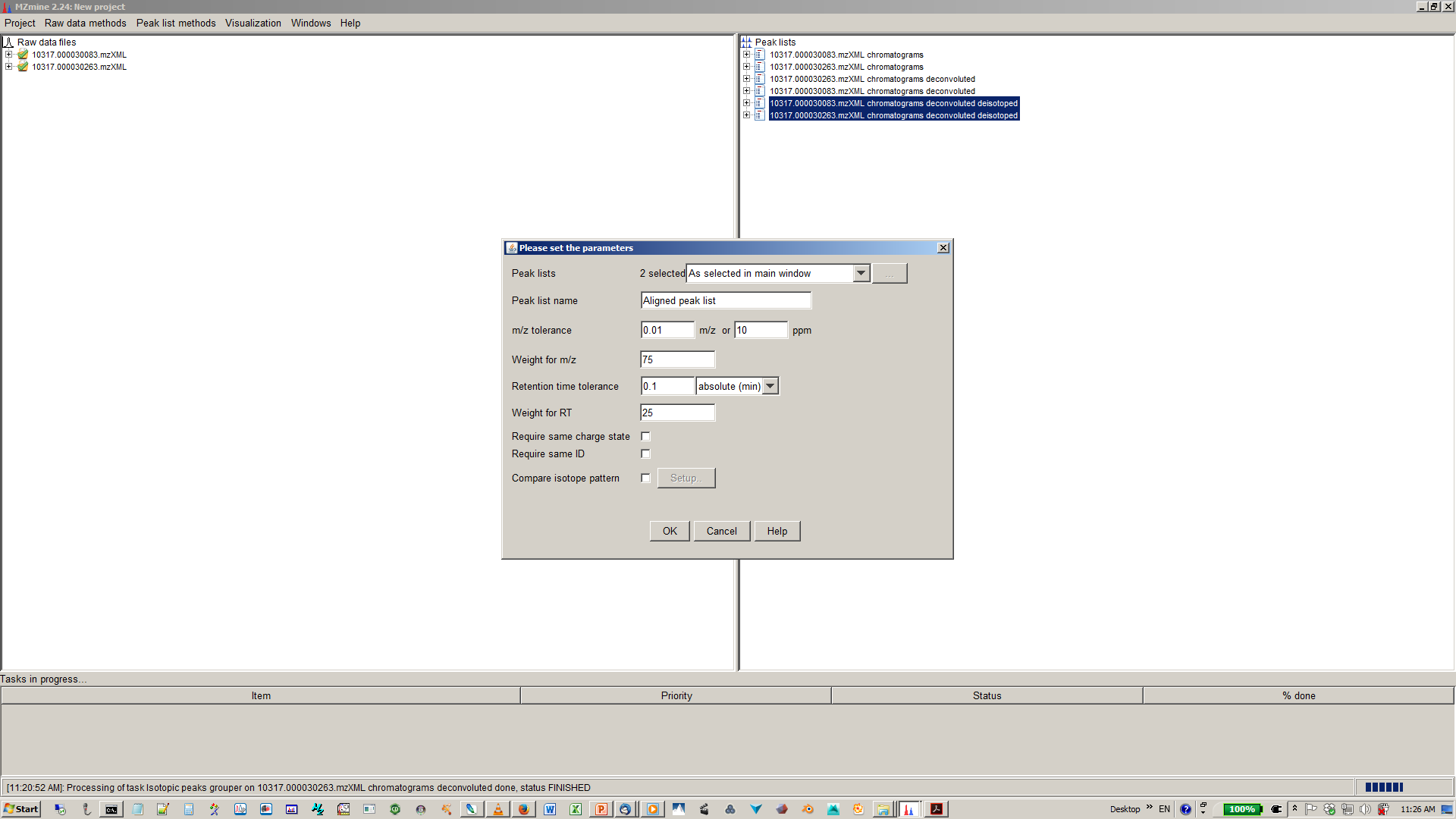
12. Specify join aligner parameters¶
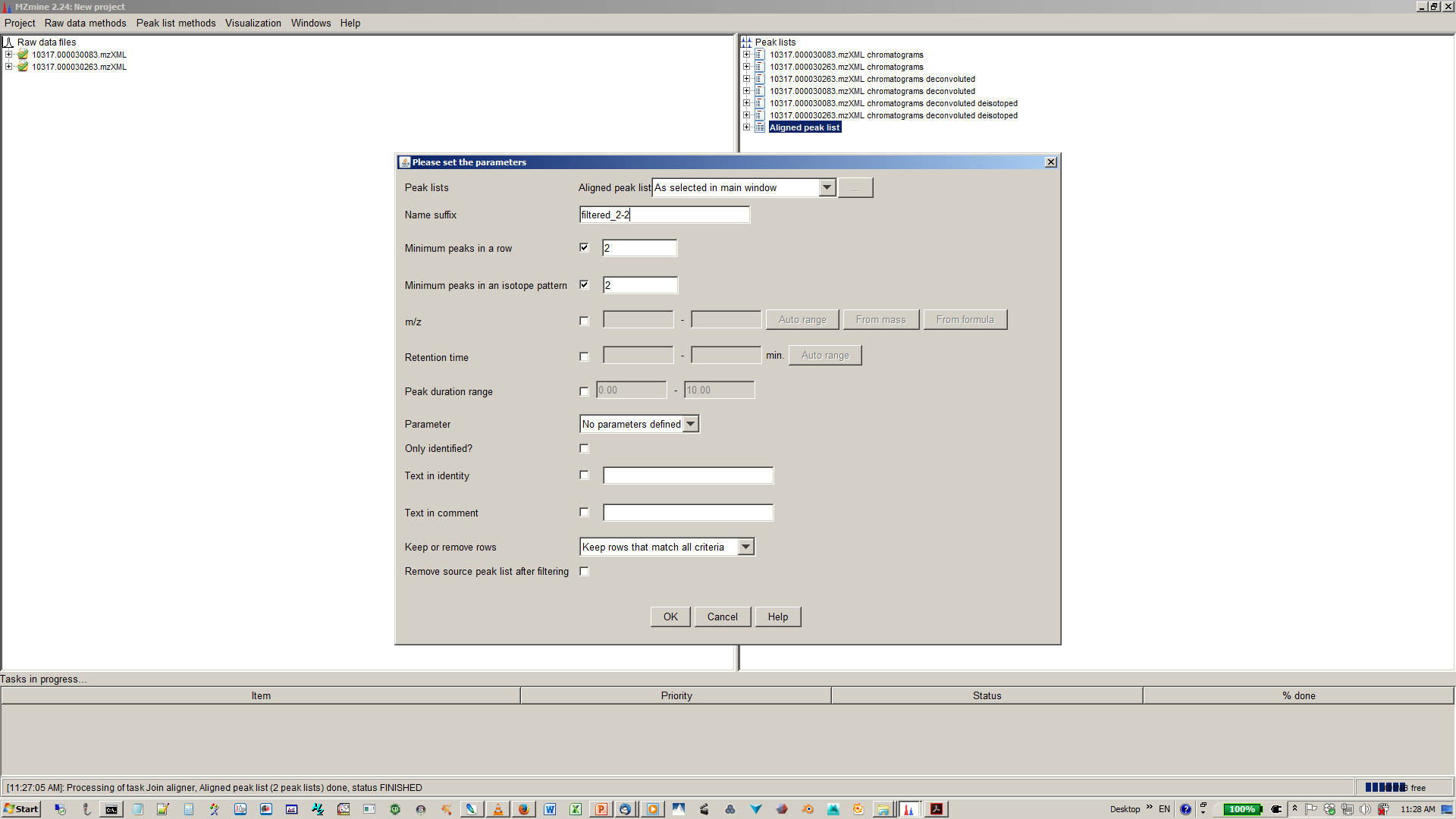
13. [optional] Filter aligned feature matrix with peak list row filter¶
14. [optional] Depending of your experimental design use n minimum peaks in a row (n should be around the number of replicates or samples you expect to be similar) and 2-3 minimum peaks per isotope pattern¶
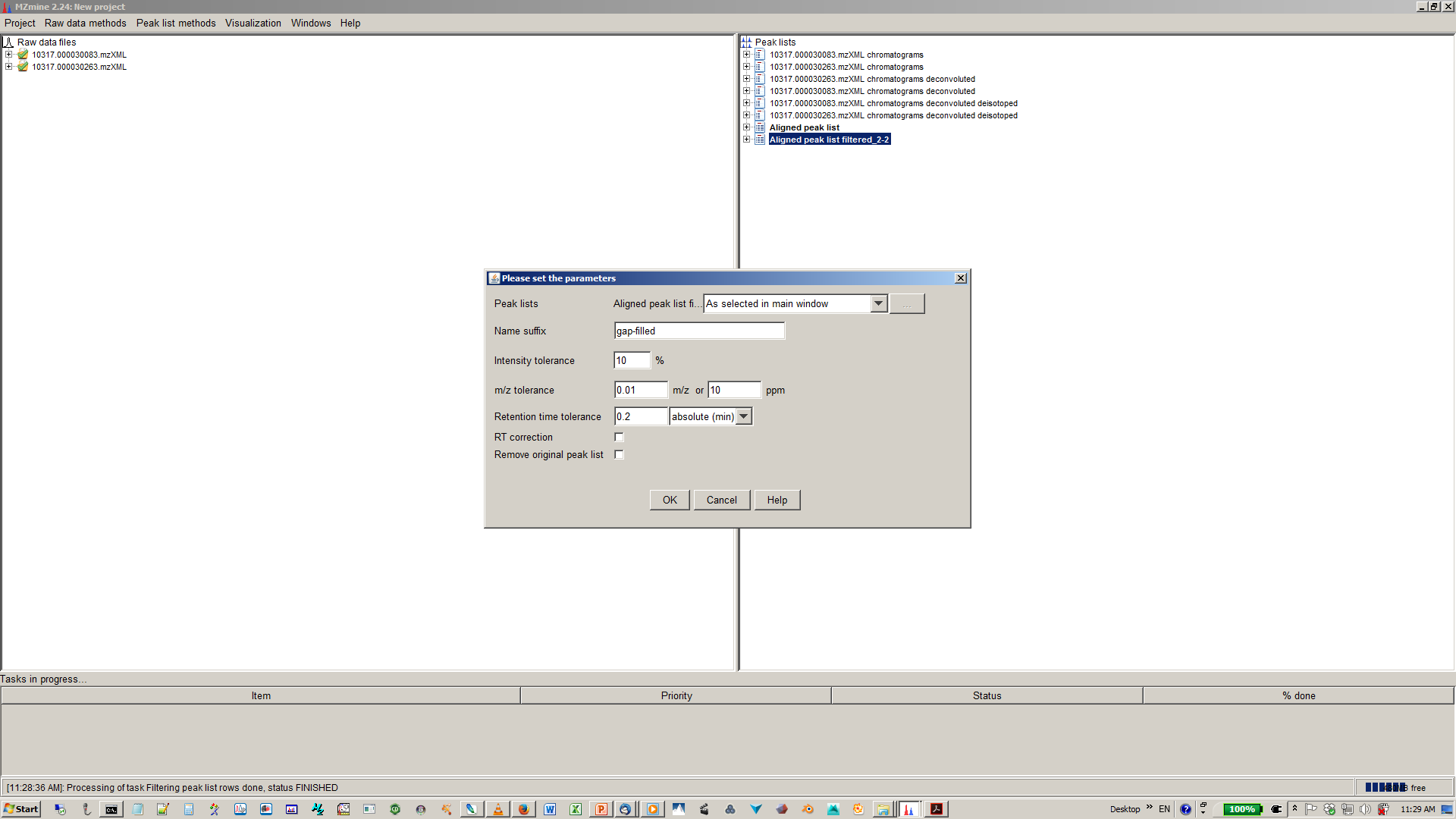
15. [optional] You gap filling the re-analyses missed peaks and fill gaps in the feature matrix¶
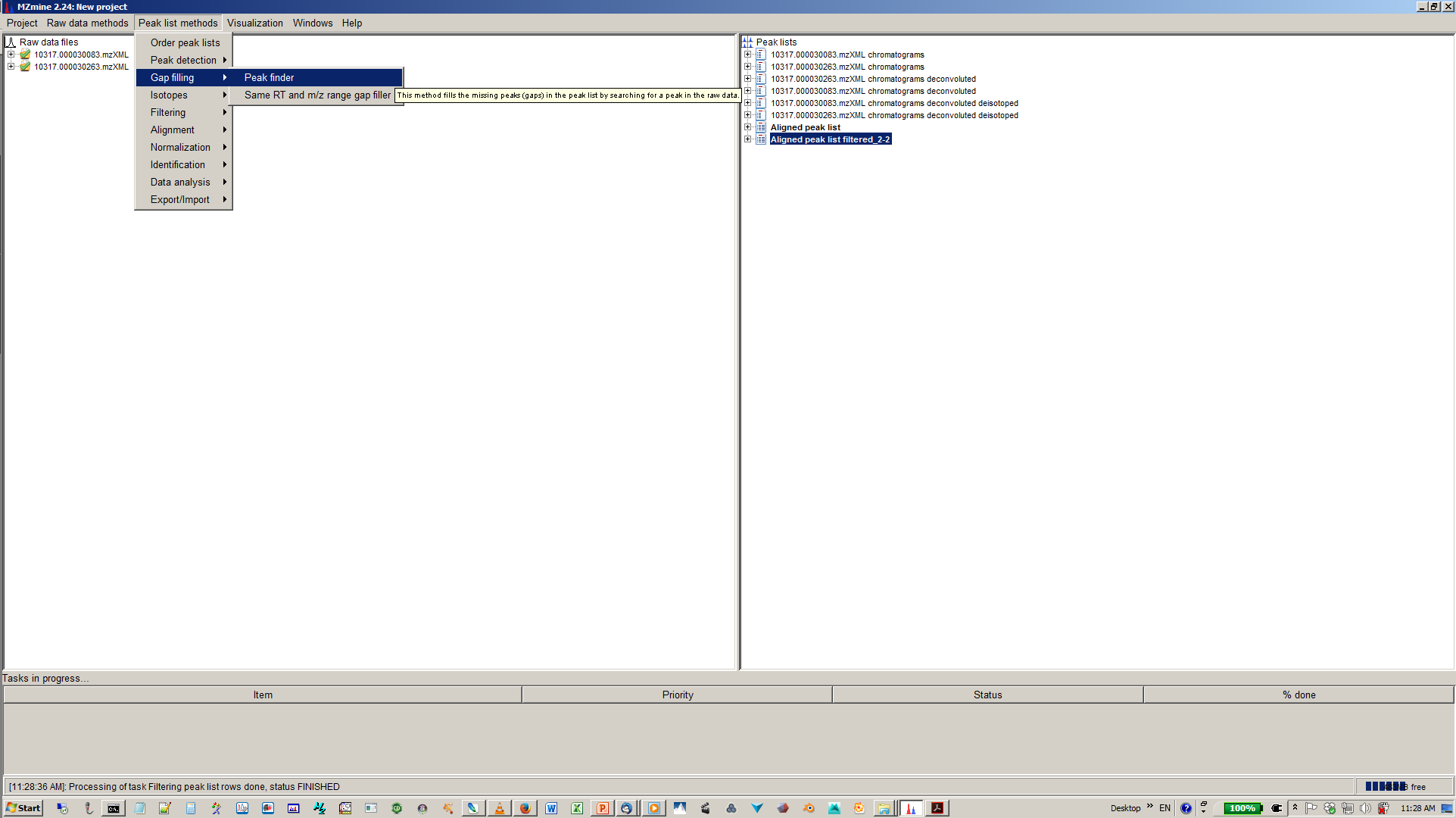
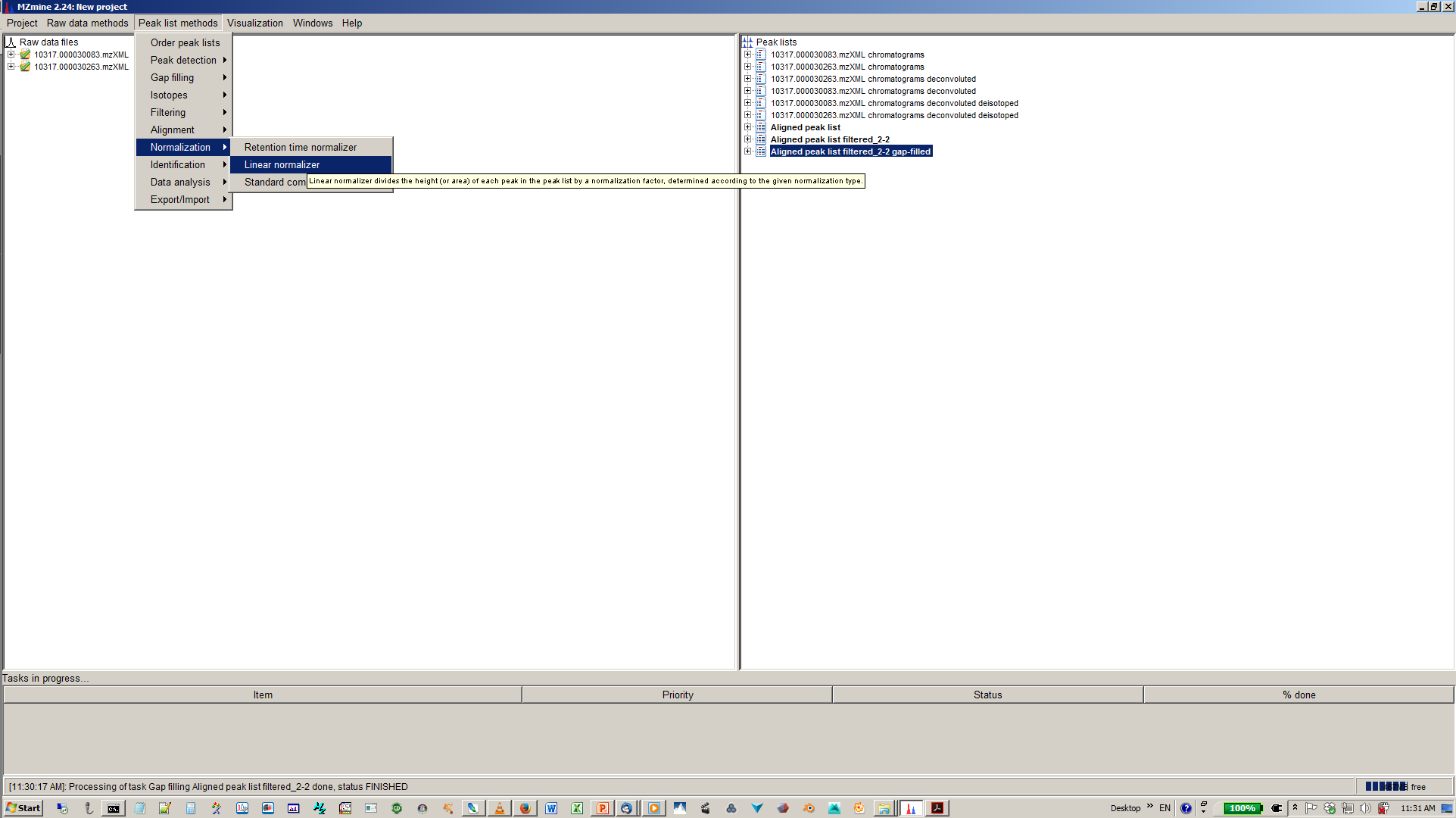
16. [optional] Depending on experimental design you can normalize your peak intensities to internal standards, TICs or total peak area.¶
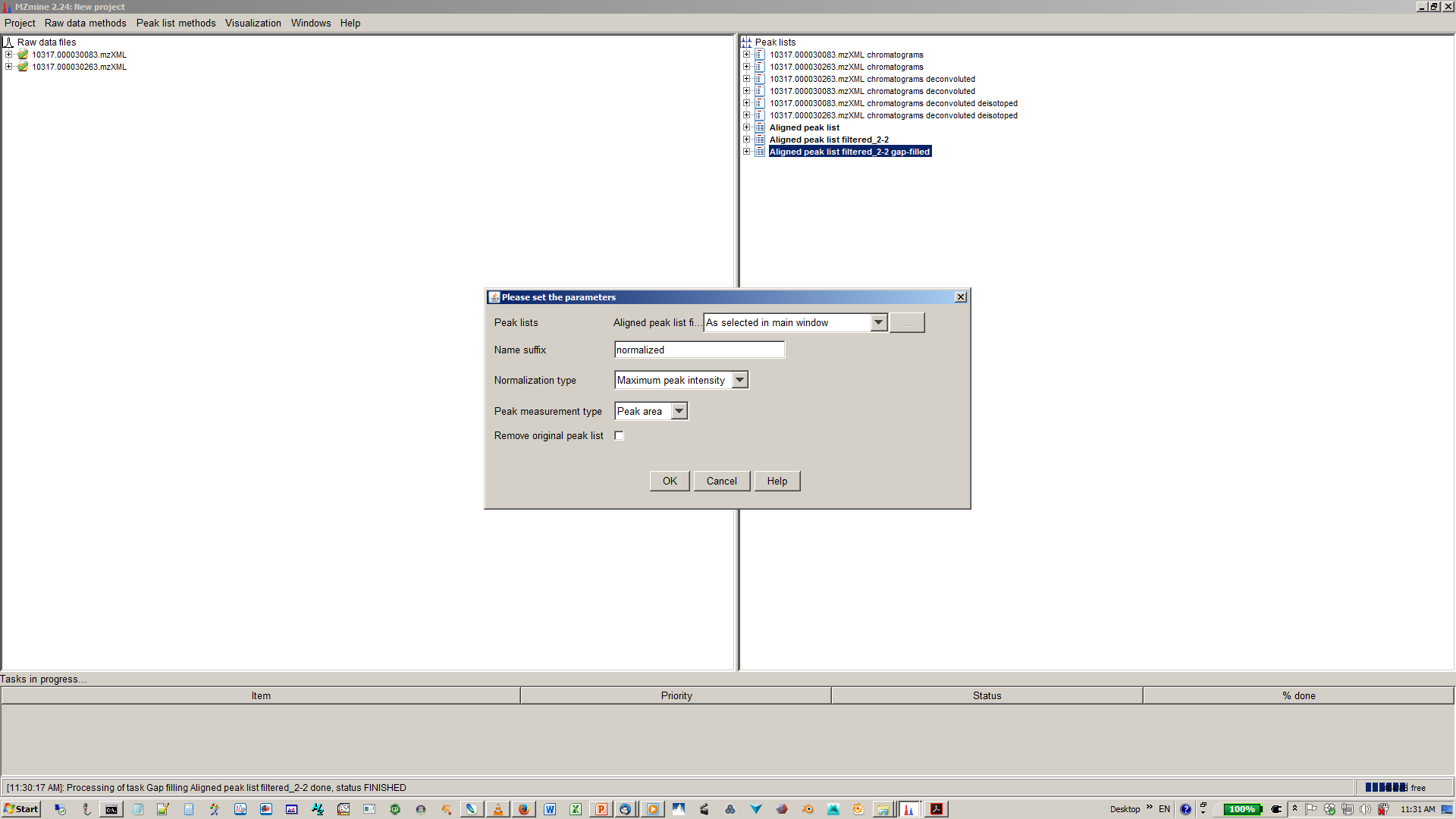
17. [optional] Specify normalization parameters¶
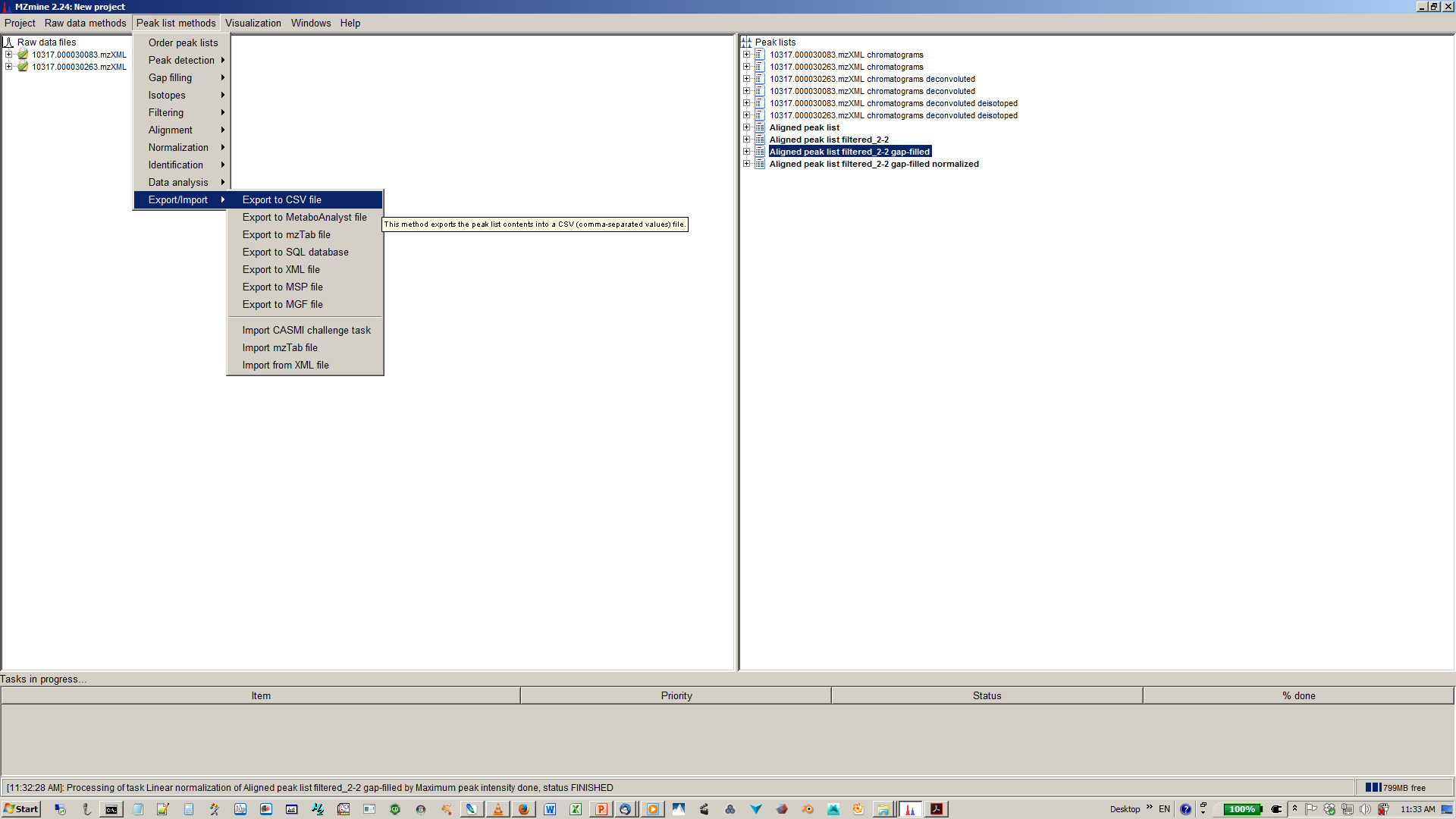
18. Export your matrix as .csv file for down stream data analysis¶
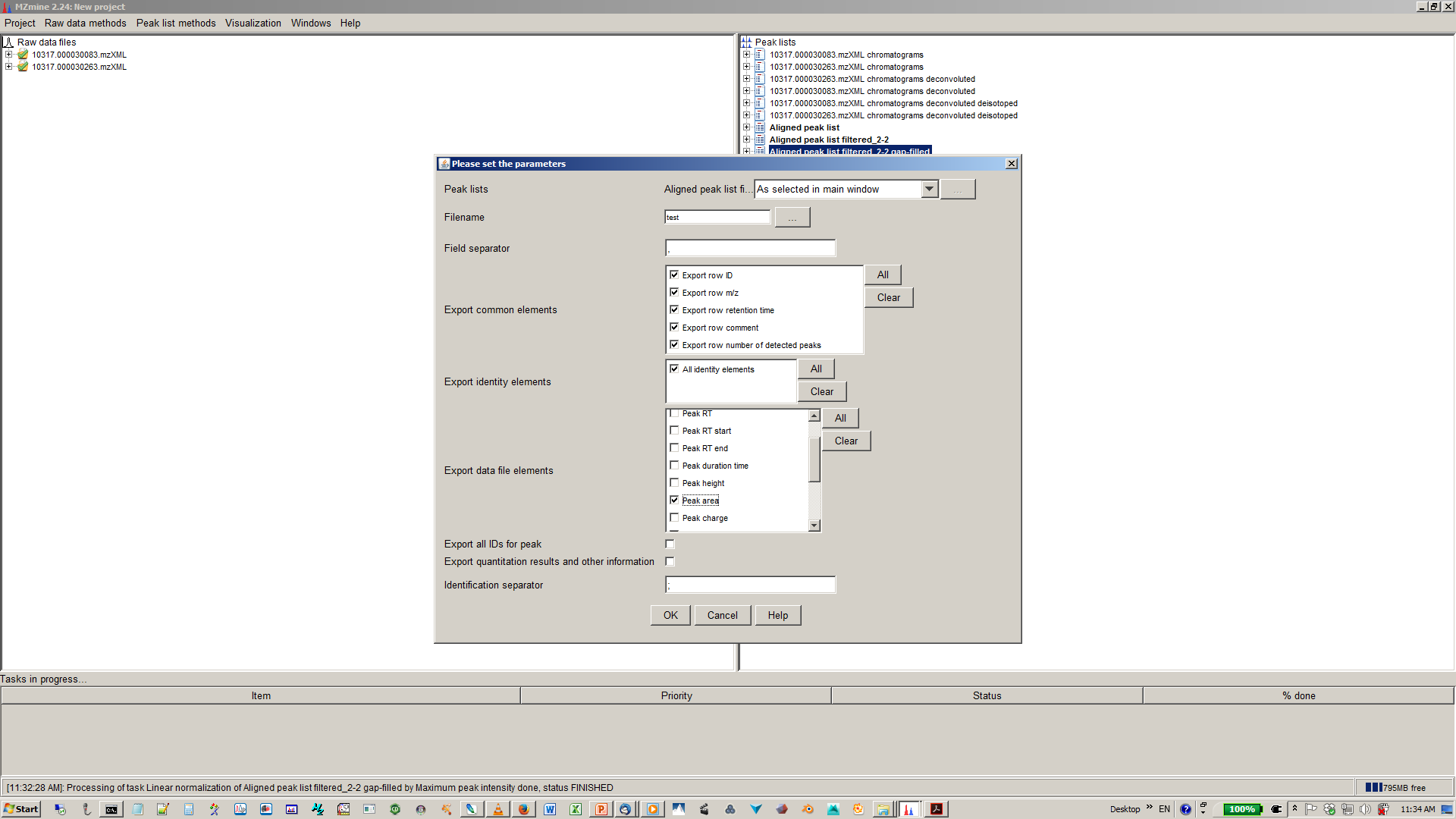
19. select file name and parameters you want to export¶
Here is also a video for MZmine 2 documentation: