16S Data Processing in Qiita¶
Now, we’ll upload some actual microbiome data to explore. To do this, we need to add the data themselves, along with some information telling Qiita about how those data were generated.
Adding a preparation template and linking it to raw data¶
Where the sample info file has the biological metadata associated with your samples, the preparation info file contains information about the specific technical steps taken to go from sample to data. Just as you might use multiple data-generation methods to get data from a single sample – for example, target gene sequencing and shotgun metagenomics – you can have multiple prep info files in a single study, associating your samples with each of these data types. You can learn more about prep info files at the Qiita documentation.
Go back to the “Upload Files” interface. In the example data, find and upload the files in the 16S folder and the file called prep_information_16S.txt.
Now you can click the “Add New Preparation” button. This will bring up the following dialogue:
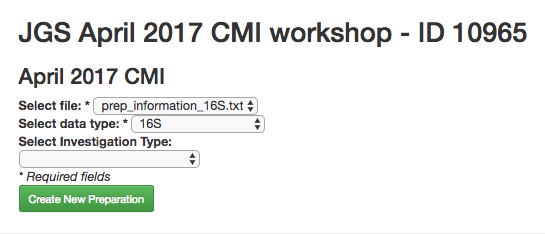
Select prep_information_16S.txt from the “Select file” dropdown, and 16S as the data type. Optionally, you can also select one of a number of investigation types that can be used to associate your data with other like studies in the database. Click “Create New Preparation”.
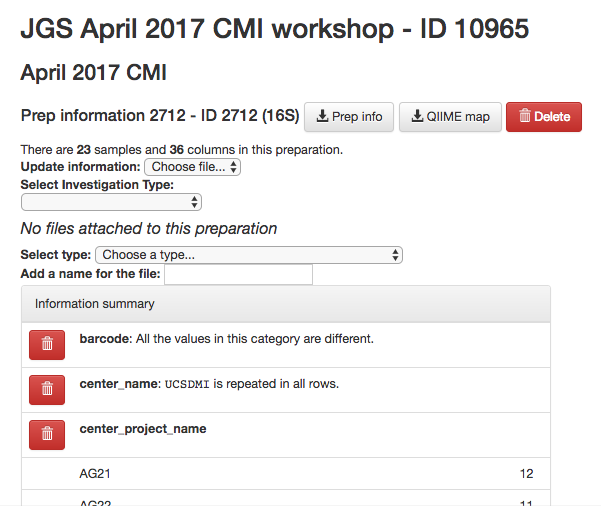
You should now see a summary of your preparation info, similar to the summary we saw of the sample info:
In addition, you should see a “16S” button appear under “Data Types” on the menu to left:
You can click this to reveal the individual prep info files of that data type that have been associated with this study:
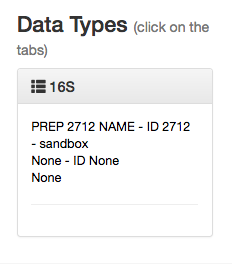
If you have multiple 16S preparations (for example, if you sequenced using several different primer sets), these would each show up as a separate entry here.
Now, you can associate the sequence data from your study with this preparation.
In the prep info dialogue, there is a dropdown menu below the words No files attached to this preparation, labeled “Select type”. Click “Choose a type” to see a list of available file types. In our case, we’ve uploaded FASTQ-formatted files per each sample in our study, so we will choose per_sample_FASTQ.
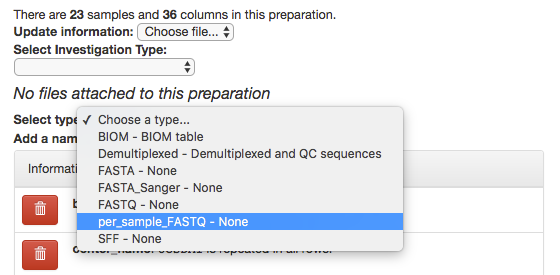
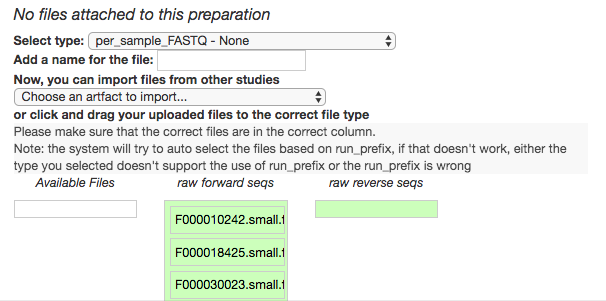
Magically, this will prompt Qiita to associate your uploaded files with the corresponding samples in your preparation info. (Our prep info file has a column named run_prefix, which associated the sample_name with the file name prefix for that particular sample.)
You should see this as a list of filenames showing up in the green raw forward seqs column below the import dropdown. You’ll want to give the set of these per-sample-FASTQ files a name (Add a name for the file), and then click “Add files” below.
That’s it! Your data are ready for processing.
Exploring the raw data¶
Click back through on your 16S preparation. Now that you’ve associated sequence files with this prep, you’ll have a Files network displayed:
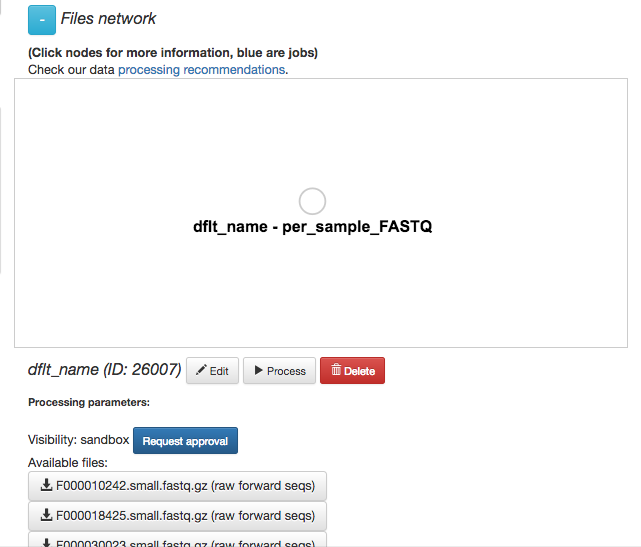
Your collection of FASTQ files for this prep are all represented by a single object in this network, currently called dflt_name. Click on the object.
Now, you’ll have a series of choices for interacting with this object. You can click “Edit” to rename the object, “Process” to perform analyses, or “Delete” to delete it. In addition, you’ll see a list of the actual files associated with this object.
Scroll to the bottom, and you’ll also see an option to generate a summary of the object.
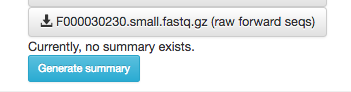
If you click this button, it will be replaced with a notification that the summary generation has been added to the processing queue.
To check on the status of the processing job, you can click the rightmost icon at the top of the screen:
This will open a dialogue that gives you information about currently running jobs, as well as jobs that failed with some sort of error.
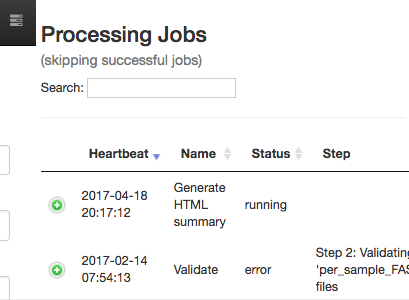
The summary generation shouldn’t take too long. When it completes, you can click back on the per_sample_FASTQ object and scroll to the bottom of the page to see a short peek at the data in each of the FASTQ files in the object. These summaries can be useful for troubleshooting.
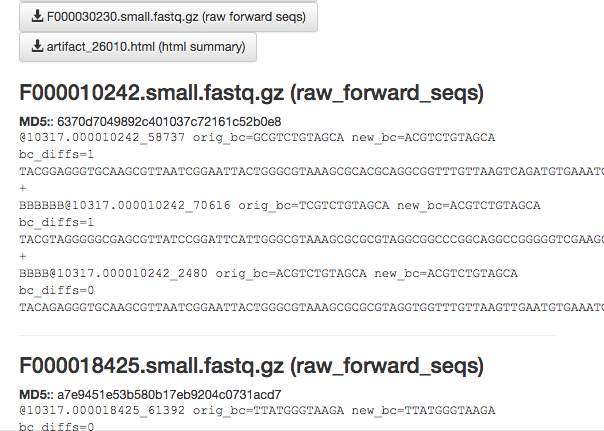
Now, we’ll process the raw data into something more interesting.
Processing 16S data¶
Scroll back up and click on the per_sample_FASTQ object, and select “Process”. This will bring you to another network visualization interface. Here, you can add processing steps to your objects.
Click again on the per_sample_FASTQ object. Below the files network, you will see an option to Choose command. Based on the type of object, this dropdown menu will give a you a list of available processing steps.
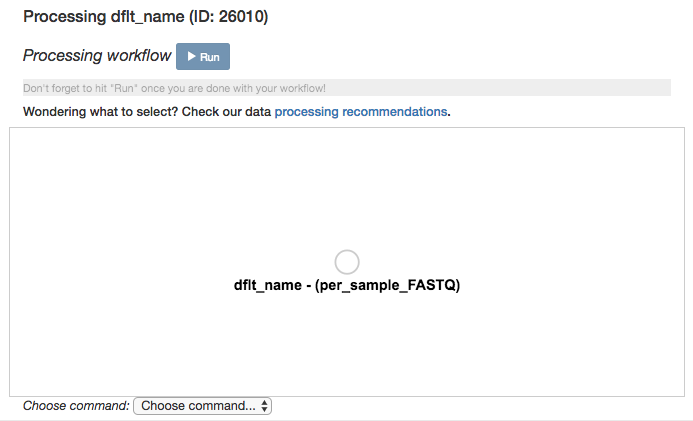
For 16S per_sample_FASTQ objects, the only available command is Split libraries FASTQ. The converts the raw FASTQ data into the file format used by Qiita for further analysis (you can read more extensively about this file type here).
Select the Split libraries FASTQ step. Now, you will be able to select the specific combination of parameters to use for this step in the Choose parameter set dropdown menu.
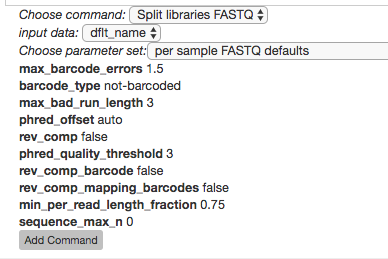
For our files, choose per sample FASTQ defaults, phred_offset 33. The specific parameter values used will be displayed below. (The other commonly used choice for data generated at the CMI is golay_12, reverse complement mapping file barcodes, reverse complement barcodes, which is what you will select if you have one set of non-demultiplexed FASTQ files (forward, reverse, and barcode) containing all of your samples.)
Click “Add Command”.
You’ll see the files network update. In addition to the original grey object, you should now see the processing command (represented in blue) and the object produced from that command (also represented in grey).
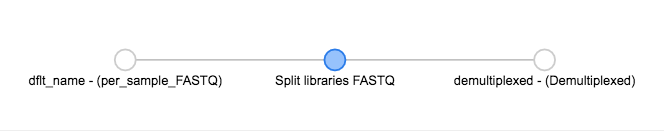
You can click on the command to see the parameters used, or on an object to perform additional steps.
Note that the command hasn’t actually been run yet! (We’ll still need to click “Run” at the top.) This allows us to add multiple processing steps to our study and then run them all together.
We’re going to process our sequences files using two different workflows. In the first, we’ll use a conventional reference-based OTU picking strategy to cluster our 16S sequences into OTUs. This approach matches each sequence to a reference database, ignoring sequences that don’t match the reference. In the second, we will use deblur, which uses an algorithm to remove sequence error, allowing us to work with unique sequences instead of clustering into OTUs. Both of these approaches work great with Qiita, because we can compare the observations between studies without having to do any sort of re-clustering!
The closed reference workflow¶
To do closed reference OTU picking, click on the demultiplexed object and select the Pick closed-reference OTUs command. We will use the default - serial parameter set for our data, which are relatively small. For a larger data set, we might want to use the parallel implementation.
By default, Qiita uses the GreenGenes 16S reference database. You can also choose to use Silva, or the Unite fungal ITS database.
Click “Add Command”, and you will see the network update:

Here you can see the blue “Pick closed-reference OTUs” command added, and that the product of the command is a BIOM-formatted OTU table.
That’s it!
The deblur workflow¶
The deblur workflow is only marginally more complex. Although you can deblur the demultiplexed sequences directly, deblur works best when all the sequences are the same length. By trimming to a particular length, we can also ensure our samples will be comparable to other samples already in the database.
Click back on the demultiplexed object. this time, select the Trimming operation. Currently, there are three trimming length options. Let’s choose Trimming 100, which trims to the first 100bp, for this run, and click “Add Command”.
Now you can see that we have the same demultiplexed object being used for two separate processing steps – closed-reference OTU picking, and trimming.
Now we can click the Trimmed Demultiplexed object and add a deblur step. Choose deblur-workflow from the Choose command dropdown, and Defaults for the parameter set. Add this command.
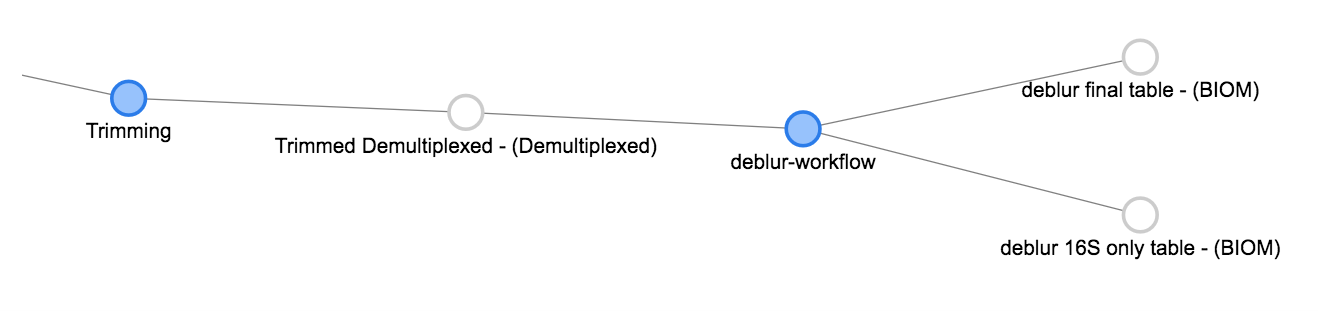
As you can see, deblur produces two BIOM-formatted OTU tables as output. The deblur 16S only table contains deblurred sequences that have been filtered to try and exclude things like organellar mitochondrial reads, while deblur final table has all the sequences.
Running the workflow¶
Now, we can see the whole set of commands and their output files:
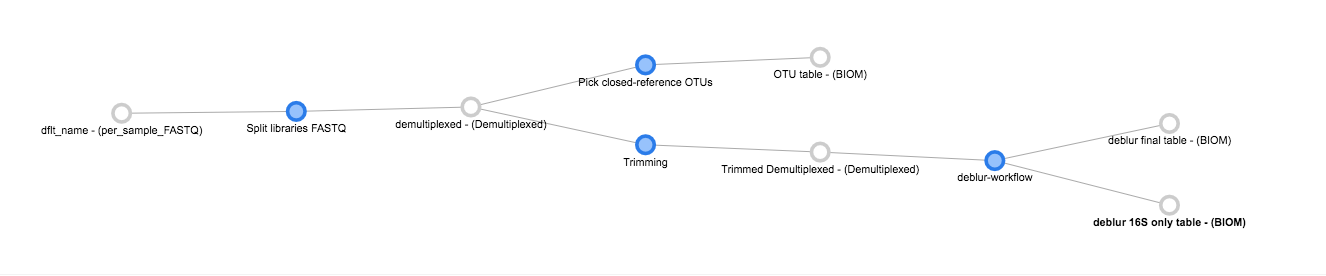
Click “Run” at the top of the screen, and Qiita will start executing all of these jobs. You’ll see a “Workflow submitted” banner at the top of your window.
As noted above, you can follow the process of your commands in the dialogue at the top right of the window.
You can also click on the objects in the prep info file network, and see status updates from the commands running on that object at the bottom of the page:

Once objects have been generated, you can generate summaries for them just as you did for the original per_sample_FASTQ object.
The summary for the demultiplexed object gives you information about the length of sequences in the object:
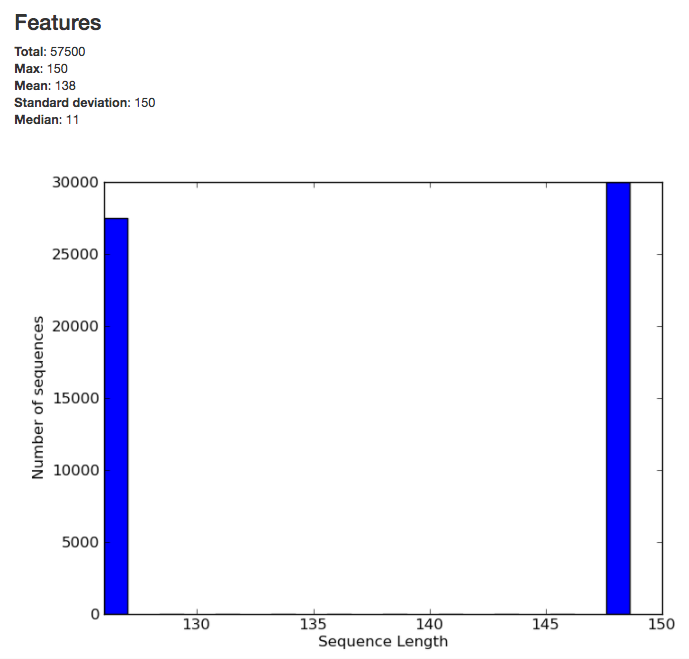
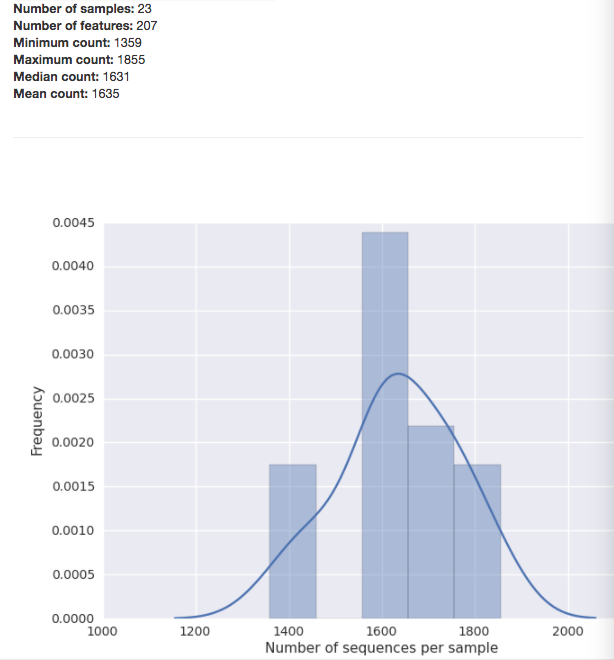
The summary for a BIOM-format OTU table gives you a histogram of the the number of sequences per sample: